NMR spektroskopi
Hvad er NMR spektroskopi og hvad bruges det til?
Kernemagnetisk Resonans (Nuclear magnetic resonance, NMR) spektroskopi er en analysemetode, som undersøger den lokale struktur omkring atomerne i en kemisk forbindelse. NMR er en ikke-destruktiv metode, så prøven ødelægges ikke ved at udføre målingen. NMR er en vigtig analysemetode indenfor mange forskellige videnskabelige områder som f.eks. kemi, mikrobiologi, miljøvidenskab samt medicinsk diagnostik (MRI). Indenfor kemien benyttes NMR ofte til at bestemme strukturen på et molekyle (f.eks. paracetamol), eller finde strukturen for en ukendt kemisk forbindelse.
I afsnittende herunder findes yderligere information om teorien bag NMR spektroskopi, samt information omkring strukturel tilordning.
Teori og praksis for NMR
NMR spektroskopi benytter, at mange atomkerner har et såkaldt spin, hvilket er et kvantemekanisk fænomen. Spin er er en egenskab i lighed med elektrisk ladning og masse for elementarpartikler - f.eks. elektroner. Hvorvidt en atomkerne har et spin, afhænger af isotopen. Alle grundstoffer har mindst én NMR-aktiv isotop. For eksempel har 1H, 2H, 13C, 14N, og 18O et kernespin hvorimod 12C og 16O isotoperne ikke har et spin. De to kan derfor ikke studeres med NMR spektroskopi.
Kernespin kan beskrives med tallet I, som kan have følgende værdier, I= ½, 1, 3/2, 2, … osv. Isotoperne 1H og 13C har begge kernespinnet I = ½, og er fokuspunktet i de følgende afsnit.
Kernespin i et magnetfelt
Et kernespin kan sammenlignes med en kompasnål. Hvis en kerne med I = ½ placeres i et magnetfelt, vil spinnet orientere sig i en bestemt retning, ligesom en kompasnål gør i jordens magnetfelt. For spin I = ½ findes kun to mulige orienteringer: En med lav energi, hvor spinnet er parallelt med det magnetiske felt (+½), og en med højere energi, hvor spinnet er rettet modsat det magnetiske felt (-½) som vist i Fig. 1.
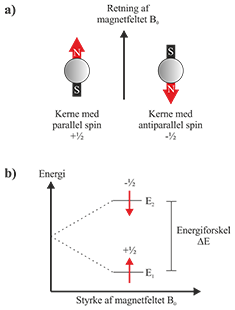
Fig. 1: a) Kernespin orienterer sig enten parallelt eller antiparallelt med et magnetfelt. b) Spinnets retning i magnetfeltet giver to energitilstande, E1 og E2.
Energiforskellen, ΔE, mellem de to tilstande er proportional med magnetfeltets styrke, og kan udtrykkes således:
ΔE = γ(h/2π)B0 (Ligning 1)
hvor B0 er styrken af magnetfeltet, h er Plancks konstant og faktoren γ kaldes det magnetogyriske forhold. En atomkerne kan overgå fra det ene energiniveau til det andet (dvs. skifte spin orientering) ved at absorbere eller udsende elektromagnetisk stråling med den samme energi som ΔE. Frekvensen, ω, af strålingen, der absorberes/udsendes, skal opfylde den såkaldte resonansligning:
ω = ΔE(2π/h) (Ligning 2)
Hver type isotop (f.eks. 1H eller 13C) har hver sin unikke γ-værdi. Dette betyder, at energiforskellen, ΔE, og dermed også frekvensen, ω, afhænger af den pågældende isotop, som undersøges.
Kemisk skift (Chemical shift)
Radiofrekvensen, som en atomkerne absorberer, angives normalt med enheden ppm (parts per million) i stedet for Hz. Dette gøres for at kunne sammenligne spektre, som er optaget med forskellig magnetstyrke. Denne ppm-værdi kaldes for kemisk skift og er angivet med δ (græsk delta).
Det kemiske skift (δ) af en atomkerne afhænger af de kemiske omgivelser atomet befinder sig i, f.eks. vil δ for 1H i en methylgruppe (R-CH3) afhænge af, hvad methylgruppen er bundet til. Ligger methylgruppen omkring 0,9 ppm i spektret, er den bundet til et andet kulstofatom. Er methylgruppen derimod bundet til ilt, vil δ-værdierne ligge i området 3,5 - 3,8 ppm. Dette er baggrunden for, at et NMR-spektrum kan bruges til at identificere stoffer.
Kemisk skift - skærmningseffekten
Hvis resonansligningen (Ligning 2) fortalte hele sandheden, ville f.eks. alle 1H-kernerne i en prøve absorbere radiobølger med nøjagtig samme frekvens, og man ville kun se ét signal i 1H NMR spektret.
Heldigvis er atomkernerne i en kemisk forbindelse omgivet af elektroner, som bevæger sig omkring kernerne. Bevægelsen af elektronerne producerer et lille magnetfelt, der skærmer atomkernerne fra det ydre magnetfelt, B0 (spektrometerets magnetfelt). Dermed er det lokale magnetfeltfelt (B), som kernerne påvirkes af, forskelligt fra B0, og kan skrives som:
B = B0(1-σ) (Ligning 3)
Værdien σ (sigma) kaldes for skærmningskonstanten og afhænger af de kemiske bindinger i molekylet. Resonansbetingelsen (Ligning 2) skal altså i virkeligheden udtrykkes som:
ω = γB0(1-σ) (Ligning 4)
Dette viser, at resonansfrekvensen er forskellig for 1H-atomer, som befinder sig i forskellige kemiske omgivelser. Kun hvis to 1H-atomer sidder i helt ens kemiske omgivelser, vil de have den samme resonansfrekvens. Herved opstår alle de signaler, vi kan se og bruge i spektret.
Et NMR-spektrometer består af en superledende magnet, en radiofrekvenssender/modtager, samt en computer, der styrer elektronikken og laver databehandling. Et NMR-spektrum optages ved at placere prøven i magnetfeltet, hvorefter en kort puls af radiobølger sendes ind i prøven. Herefter optager spektrometret hvilke radiobølgefrekvenser, som absorberes af prøven.
De fleste kommercielle NMR spektrometre har en magnetstyrke mellem 1,4 Tesla til 25,9 Tesla. For isotopen 1H svarer det til frekvenser i området 60 MHz til 1,1 GHz. De dyreste NMR speketrometre har de kraftigste magneter. I alle tilfælde ligger strålingen i radiobølgeområdet af det elektromagnetiske spektrum. Bemærk at dette svarer til en meget lav energi i forhold til de infrarøde eller synlige dele af det elektromagnetiske spektrum.
Kemisk skift område
De kemiske skift (δ) værdier for 1H ligger oftest i området 0 ppm til 14 ppm. I Fig. 1 er δ-værdierne for forskellige kemiske bindinger angivet, og kan benyttes som hjælp til tilordning af 1H NMR spektre.
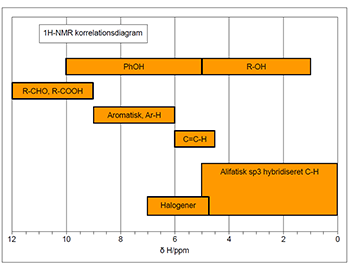
Figur 1: Oversigt over kemisk skift områder i 1H-NMR
Intensiteter
Intensiteterne af de observerede 1H-signaler er et direkte mål for hvor mange 1H-kerner, der giver anledning til signalerne. Som et eksempel benyttes ethanol (CH3CH2OH), her vil man se tre forskellige signaler, hvis intensitet har forholdet 3:2:1. Dermed kan intensitets-forholdene bruges til at bestemme mol-forholdene i en blanding af forskellige stoffer. Intensiteterne bestemmes ud fra arealet under signalerne (integralet) og findes nemt med databehandlingssoftwaren.
For at illustrere dette ses NMR-spektret af 2,2-dimethyl-propanol (Fig. 2), hvor man kan se toppene for de tre methylgrupper og ethylgruppen. Forholdet mellem toppene måles til 2:9 (angivet under toppene), hvilket betyder at der i linjen ved δ = 3,3 ppm vil være 2 protoner for hver 9 protoner i linjen ved 0,97 ppm.
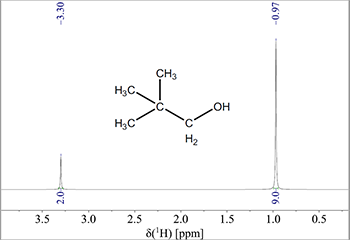
Figur 2: Delspektrum af 2,2-dimethyl-propanol, hvor integralet er angivet under toppene.
J-kobling: Introduktion
Kun meget sjældent vil et NMR-spektrum være så simpelt som hidtil beskrevet. Eksperimentelt observeres langt flere linjer i spektret, end hvad der forventes ud fra analysen af kemisk skift. Årsagen er, at atomkernernes spin påvirker hinanden. Da kernernes spin producerer små magnetfelter, kan de således påvirke det kemiske skift for de 1H, som sidder i nærheden. Dette kaldes for J-kobling, og forskellige eksempler introduceres i følgende underafsnit.
J-kobling: 1H-1H kobling
Lad os betragte et molekylfragment, -CH-CH-. Dette fragment har kun to protoner, som vi kalder HA og HB:
Der er kun 3 bindinger mellem HA og HB, og de sidder derfor så tæt på hinanden i molekylet at de kan mærke hinandens tilstedeværelse. I en stofprøve med dette molekylefragment, vil HB i nogle af molekylerne have spin +½ (X-type molekyle), og i andre molekyler vil HB have spin -½ (Y-type molekyle). Fig. 3 illustrerer disse to typer af molekyler. De magnetiske omgivelser omkring proton HA afhænger af om proton HB er i +½ eller -½ spin-tilstanden (HA siges at koble med HB). Derfor absorberer proton HA elektromagnetiskstråling ved lidt forskellig frekvens i de to tilfælde, og dermed afhænger det kemiske skift for proton HA af retningen på spinnet af proton HB. Faktisk er proton HA i X-type molekyler lidt mindre skærmet end proton HA i Y-type molekyler. Det betyder at det samlede spektrum af proton HA, der er summen af både spektret fra X-type og Y-type molekyler, vil bestå af to linjer der er forskudt lidt fra hinanden. Nøjagtig samme situation beskrives for proton HB. Denne proton giver også to linjer, én for molekyler med proton HA i +½ spin-tilstand og én for molekyler med proton HA i -½ spintilstand. Det betyder at 1H NMR spektret af molekyle-fragmentet vil bestå af 2 dubletter som vist på Fig. 3.
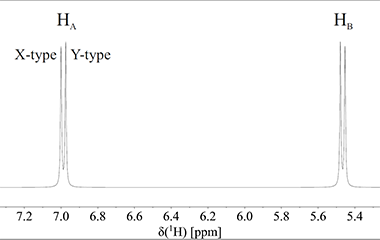
Figur 3: Spektrum for to protoner, der kobler med hinanden og dermed splitter op i to dubletter.
J-kobling: 1H-1H2 kobling
Hvis vi i stedet havde betragtet molekylfragmentet -CH-CH2- kan man på tilsvarende måde forestille sig at CH2-protonerne vil give en dublet fordi CH-protonen enten kan have spin +½ eller spin -½. Derimod vil spektret af CH-protonen give tre linjer fordi hver af de to CH2 protoner kan have enten spin +½ (↑) eller spin -½ (↓). De giver tre mulige spin tilstande for de to CH2-protoner:
|
Tilstand 1 |
Tilstand 2 |
Tilstand 3 |
|
↓↓ |
↓↑ |
↑↑ |
|
|
↑↓ |
|
I tilfælde 2 ophæver de to spin hinandens virkning på CH-protonen, og vi får en linje ved kemisk værdien (altså svarende til ingen J-kobling). I de to andre tilfælde forstærker de to spin hinandens virkning. Det betyder at vi får en triplet med intensitetsforholdene 1:2:1 som vist på Fig. 4.
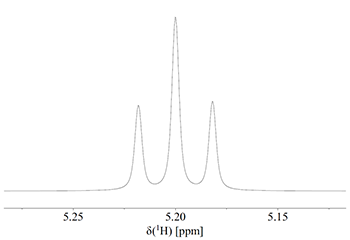
Figur 4: Triplet med intensitet 1:2:1.
J-kobling: n+1 regel
Generelt gælder reglen: hvis én 1H sidder i nærheden af n andre 1H vil signalet splitte op i (n + 1)-antal linjer. Afstanden mellem linjerne i opsplitningerne kaldes spin-spin koblingskonstanten J og er angivet i Hz Intensitetsforholdet i opsplitningen er bestemt af Pascals trekant:
|
|
Opsplitning samt intensitet |
||||||||||||
|
n=0 |
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
n=1 |
|
|
|
|
|
1 |
|
1 |
|
|
|
|
|
|
n=2 |
|
|
|
|
1 |
|
2 |
|
1 |
|
|
|
|
|
n=3 |
|
|
|
1 |
|
3 |
|
3 |
|
1 |
|
|
|
|
n=4 |
|
|
1 |
|
4 |
|
6 |
|
4 |
|
1 |
|
|
|
n=5 |
|
1 |
|
5 |
|
10 |
|
10 |
|
5 |
|
1 |
|
|
n=6 |
1 |
|
6 |
|
15 |
|
20 |
|
15 |
|
6 |
|
1 |
Dog gælder n+1 reglen kun når der er én koblingskonstant, J, tilstede. Hvis en 1H kobler med to andre 1H med forskellige koblingskonstanter bliver spektret mere kompliceret. Man kan med god tilnærmelse regne med, at i alifatiske forbindelser (-CH-, -CH2-, -CH3) er koblingskonstanten mellem to 1H der sidder mere end tre bindinger fra hinanden forsvindende lille. I forbindelser, der indeholder dobbelt- eller trippelbindinger (C=C, C≡C), eller i aromatiske forbindelser (f.eks. benzenringe), kan man derimod normalt se små koblinger op til fire og fem bindinger væk.
Ækvivalente 1H vil ikke vise spin-spin kobling med hinanden. F.eks. vil en CH2-resonans ikke splitte op fordi der ikke ses en kobling mellem de to protoner (men den vil naturligvis splitte op på grund af kobling til nabogrupper som f.eks. -CH3). Som et eksempel på dette ses nedenfor spektret af 1-brom-3-chlorpropan i Fig. 5.
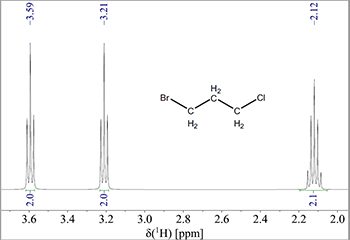
Figur 5: NMR-spektrum af 1-brom-3-chloropropan.
13C er den eneste naturligt forekommende C-isotop med et kernespin (I = ½), og er dermed også den eneste NMR aktive C-isotop. Desværre er den naturlige forekomst af 13C ca. 1.1 %. Derfor er 13C NMR spektroskopi betydeligt vanskeligere end 1H spektroskopi, da den naturlige forekomst af 1H er 99.98 %.
Derfor bringes andre metoder i anvendelse, som muliggør 13C NMR spektroskopi. Dog forsimpler disse metoder 13C spektret. Det betyder at J-koblinger ikke observeres i 13C NMR spektroskopi, og 13C intensiteterne ikke afhænger af det tilsvarende antal kulstofatomer i prøven, som det ellers ses i 1H NMR spektroskopi.
Kemisk skift område
De kemiske skift værdier for 13C ligger oftest i området 0 ppm til 250 ppm, hvilket er mere end 10 gange større end for 1H. I Fig. 1 er δ-værdierne for forskellige kemiske bindinger angivet, og kan benyttes som hjælp til tilordning af 13C-NMR spektre.
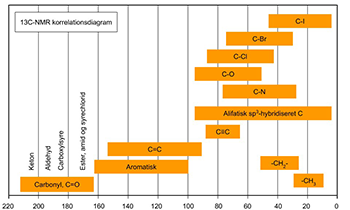
Figur 1: Oversigt over kemisk skift for 13C-NMR
13C-NMR spektre af carbonylgrupper (C=O), cyanid (C≡N), carboxylsyre grupper (COOH) o.l. giver alle karakteristiske δ-værdier. Deres signaler vil ofte være svage, idet disse kulstofatomer er uden direkte bindinger til 1H atomer.
I grove træk minder 13C signalernes placering om den rækkefølge og substituent-afhængighed, som gælder for 1H. F.eks. hvis et 1H-spektrum har linjer, der ligger højt i spektret, vil signalerne for 13C bundet til disse protoner også ligge højt i 13C spektret.
- D. L. Pavia, G. M. Lampman, G. S. Kriz and J. R. Vyvyan, Introduction to Spectroscopy, Cengage Learning, 2015.
- H. Friebolin, Basic One- and Two-Dimensional NMR Spectroscopy, Wiley, 5 edn., 2010.