Highlights
Adipocyte gene expression in obesity - insights gained and challenges ahead
Lasse K. Markussen, Susanne Mandrup (2023). Curr Opin Genet Dev. https://doi.org/10.1016/j.gde.2023.102060
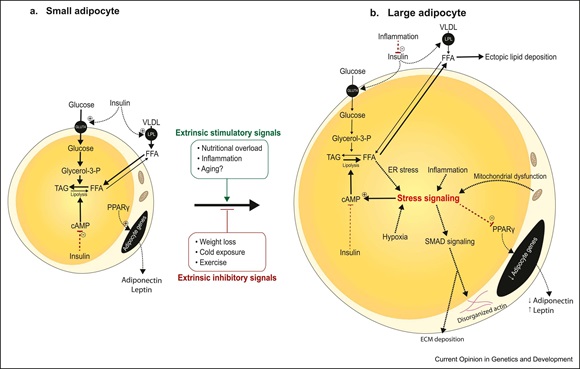
White adipocytes possess extraordinary plasticity with an unparalleled capacity to expand in size with nutritional overload. Several lines of evidence indicate that limitations to this plasticity, as found in both lipodystrophy and obesity, drive several of the comorbidities of these disease, thereby underscoring the need to understand the mechanisms of healthy and unhealthy adipose expansion. Recent single-cell technologies and studies of isolated adipocytes have allowed researchers to gain insight into the molecular mechanisms of adipocyte plasticity. Here, we review current insight into the effect of nutritional overload on white adipocyte gene expression and function. We review the role of adipocyte size and heterogeneity and discuss the challenges and future directions.
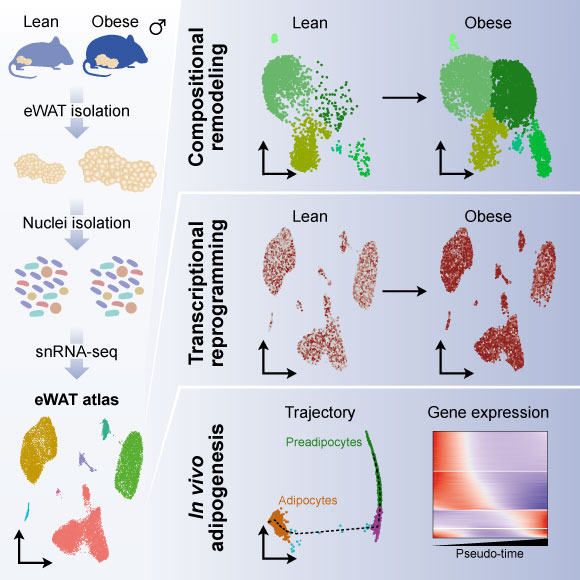
Single-cell-resolved transcriptional dynamics of human subcutaneous adipose tissue during lifestyle- and bariatric surgery-induced weight loss
Anne Loft, Rasmus Rydbirk, Ellen Gammelmark Klinggaard, Elvira Laila Van Hauwaert, Charlotte Wilhelmina Wernberg, Andreas Fønss Møller, Trine Vestergaard Dam, Mohamed Nabil Hassan, Babukrishna Maniyadath, Ronni Nielsen, Aleksander Krag, Joanna Kalucka, Søren Fisker Schmidt, Mette Enok Munk Lauridsen, Jesper Grud Skat Madsen, Susanne Mandrup (2026). Nat Metab. https://doi.org/10.1038/s42255-025-01433-4
Human white adipose tissue undergoes major remodelling during sustained weight gain that may compromise tissue function and drive cardiometabolic comorbidities. Although weight loss reverses many of these complications, the cellular and molecular adaptations of adipose tissue to different weight loss interventions are poorly understood. Here we show how abdominal subcutaneous adipose tissue (SAT) in men and women with severe obesity adapts to modest lifestyle-induced (8-10%) weight loss followed by substantial bariatric surgery-induced (20-45%) weight loss, using single-nucleus RNA sequencing (snRNA-seq) combined with bulk RNA-seq, and three-dimensional light-sheet fluorescence microscopy. To enable interactive exploration, all snRNA-seq data are available in a browsable format on the Single Cell Portal ( SCP2849 ). Lifestyle-induced weight loss activated proadipogenic gene programmes in progenitor cells, indicating early beneficial effects on SAT. Subsequent surgery-induced weight loss drove profound compositional and transcriptional remodelling of SAT, including increased vascularization and marked reduction of myeloid cell populations. Collectively, our study indicates that following major and sustained weight loss, SAT from individuals with severe obesity has the capacity to return to a state comparable to that observed in lean individuals.

Extensive enhancer crosstalk controls PPARG2 activation during adipogenesis
Anna Cetnarowska, Mette Hyldahl, Marcus Nygård, Hesam Dashti, Bo Vagner Hansen, Laura Kristine Holm, Kaja Madsen, Maria Stahl Madsen, Vallari Shukla, Esra Durmaz Mitchell, Alexander Rauch, Jesper Grud Skat Madsen, Melina Claussnitzer, Susanne Mandrup (2026). Nat Commun. https://doi.org/10.1038/s41467-026-70401-7
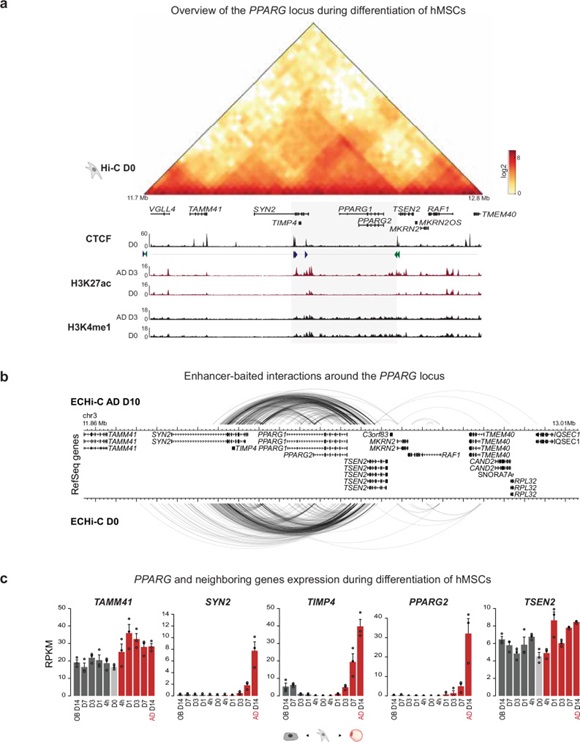
Transcriptional master regulators drive cell fate transitions. Peroxisome proliferator-activated receptor γ (PPARγ) is the master regulator of adipogenesis, and its expression must therefore be tightly regulated and efficiently induced in response to adipogenic cues. Here we decipher the regulatory mechanisms of the highly connected enhancer community driving activation of the PPARG locus during adipocyte differentiation of human mesenchymal stem cells. By systematically deleting nine individual enhancers, spanning upstream, promoter-proximal, and downstream super-enhancer constituents, we demonstrate elaborate enhancer crosstalk in cis involving stabilization of C/EBPβ recruitment prior to chromatin remodeling. We show that the super-enhancer constituent E + 102 plays a dual role in cis crosstalk and feedback activation and is obligate for activation of PPARG expression. Non-coding genetic variants associated with cardiometabolic traits and predicted to regulate PPARG expression map to E + 102 and other essential enhancers in the community, thereby supporting the importance of these enhancers in human physiology and disease.
Towards a consensus atlas of human and mouse adipose tissue at singe-celle resolution
A. Loft, M. P. Emont , A. Weinstock , A. Divoux, A. Ghosh, A. Wagner, A. V. Hertzel, B. Maniyadath, B. Deplancke, B. Liu, C. Scheele, C. Lumeng, C. Ding, C. Ma, C. Wolfrum , C. Strieder-Barboza, C. Li, D. D. Truong, D. A. Bernlohr, E. Stener-Victorin, E. E. Kershaw, E. Yeger-Lotem, F. Shamsi, H. X. Hui, H. Camara, J. Zhong, J. Kalucka, J. A. Ludwig, J. A. Semon, J. Jalkanen, K. L. Whytock, K. D. Dumont, L. M. Sparks, L. A. Muir, L. Fang, L. Massier, L. R. Saraiva, M. D. Beyer, M. G. Jeschke, M. A. Mori, M. Boroni, M. J. Walsh, M-E. Patti, M. D. Lynes, M. Blüher, M. Rydén, N. Hamda, N. L. Solimini, N. Mejhert, P. Gao, R. K. Gupta, R. Murphy, Sa. Pirouzpanah, S. Corvera, S. Tang, S. K. Das, S. F. Schmidt, T. Zhang, T. M. Nelson, T. E. O’Sullivan, V. Efthymiou, W. Wang, Y. Tong, Y-H. Tseng, S. Mandrup & E. D. Rosen (2025). Nature Metabolism. https://doi.org/10.1038/s42255-025-01296-9
Adipose tissue (AT) is a complex connective tissue with a high relative proportion of adipocytes, which are specialized cells with the ability to store lipids in large droplets. AT is found in multiple discrete depots throughout the body, where it serves as the primary repository for excess calories. In addition, AT has an important role in functions as diverse as insulation, immunity and regulation of metabolic homeostasis. The Human Cell Atlas Adipose Bionetwork was established to support the generation of single-cell atlases of human AT as well as the development of unified approaches and consensus for cell annotation. Here, we provide a first roadmap from this bionetwork, including our suggested cell annotations for humans and mice, with the aim of describing the state of the field and providing guidelines for the production, analysis, interpretation and presentation of AT single-cell data.
Lipolysis regulates major transcriptional programs in brown adipocytes
L. K. Markussen, E. A. Rondini, O. S. Johansen, J. G. S. Madsen, E. G. Sustarsic, A-B. Marcher, J. B. Hansen, Z. Gerhart-Hines, J. G. Granneman, S. Mandrup (2022). Nat Commun. https://doi.org/10.1038/s41467-022-31525-8
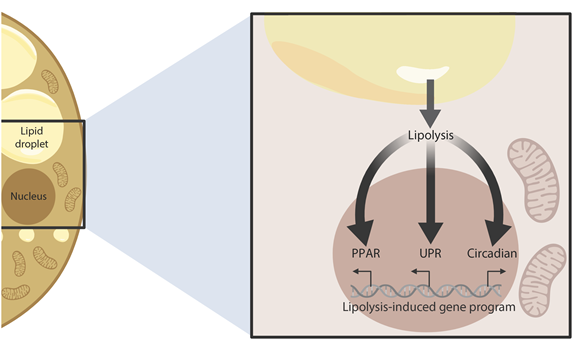
β-Adrenergic signaling is a core regulator of brown adipocyte function stimulating both lipolysis and transcription of thermogenic genes, thereby expanding the capacity for oxidative metabolism. We have used pharmacological inhibitors and a direct activator of lipolysis to acutely modulate the activity of lipases, thereby enabling us to uncover lipolysis-dependent signaling pathways downstream of β-adrenergic signaling in cultured brown adipocytes. Here we show that induction of lipolysis leads to acute induction of several gene programs and is required for transcriptional regulation by β-adrenergic signals. Using machine-learning algorithms to infer causal transcription factors, we show that PPARs are key mediators of lipolysis-induced activation of genes involved in lipid metabolism and thermogenesis. Importantly, however, lipolysis also activates the unfolded protein response and regulates the core circadian transcriptional machinery independently of PPARs. Our results demonstrate that lipolysis generates important metabolic signals that exert profound pleiotropic effects on transcription and function of cultured brown adipocytes.
Plasticity of Epididymal Adipose Tissue in Response to Diet-Induced Obesity at Single-Nucleus Resolution
A. K. Sávári, E. L. Van Hauwaert, L. K. Markussen, E. Gammelmark, A-B. Marcher, M. F. Ebbesen, R. Nielsen, J. R. Brewer, J. G. S. Madsen, S. Mandrup (2021). Cell Metab. https://doi.org/10.1016/j.cmet.2020.12.004
Adipose tissues display a remarkable ability to adapt to the dietary status. Here, we have applied single-nucleus RNA-seq to map the plasticity of mouse epididymal white adipose tissue at single-nucleus resolution in response to high-fat-diet-induced obesity. The single-nucleus approach allowed us to recover all major cell types and to reveal distinct transcriptional stages along the entire adipogenic trajectory from preadipocyte commitment to mature adipocytes. We demonstrate the existence of different adipocyte subpopulations and show that obesity leads to disappearance of the lipogenic subpopulation and increased abundance of the stressed lipid-scavenging subpopulation. Moreover, obesity is associated with major changes in the abundance and gene expression of other cell populations, including a dramatic increase in lipid-handling genes in macrophages at the expense of macrophage-specific genes. The data provide a powerful resource for future hypothesis-driven investigations of the mechanisms of adipocyte differentiation and adipose tissue plasticity. Read more here.
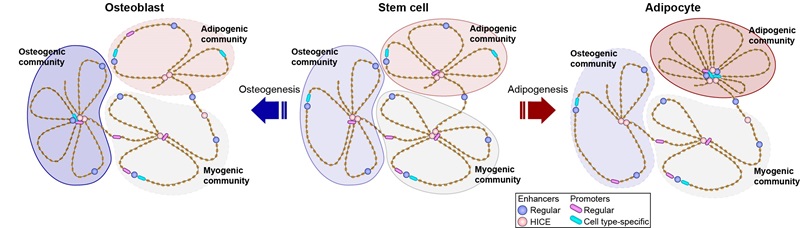
Highly interconnected enhancer communities control lineage-determining genes in human mesenchymal stem cells
J. G. S. Madsen, M. S. Madsen, A. Rauch, S. Traynor, E. L. V. Hauwaert, A. K. Haakonsson, B. M. Javierre, M. Hyldahl, P. Fraser & S. Mandrup (2020). Nature Genetics. https://doi.org/10.1038/s41588-020-0709-z
Adipocyte differentiation is driven by waves of transcriptional regulators that reprogram the enhancer landscape and change the wiring of the promoter interactome. Here, we use high-throughput chromosome conformation enhancer capture to interrogate the role of enhancer-to-enhancer interactions during differentiation of human mesenchymal stem cells. We find that enhancers form an elaborate network that is dynamic during differentiation and coupled with changes in enhancer activity. Transcription factors (TFs) at baited enhancers amplify TF binding at target enhancers, a phenomenon we term cross-interaction stabilization of TFs. Moreover, highly interconnected enhancers (HICE) act as integration hubs orchestrating differentiation by the formation of three-dimensional enhancer communities, inside which, HICE, and other enhancers, converge on phenotypically important gene promoters. Collectively, these results indicate that enhancer interactions play a key role in the regulation of enhancer function, and that HICE are important for both signal integration and compartmentalization of the genome.
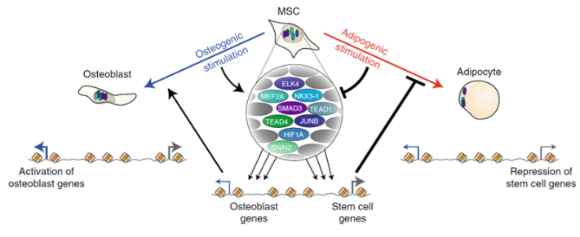
Osteogenesis depends on commissioning of a network of stem cell transcription factors that act as repressors of adipogenesis
Alexander Rauch, Anders K. Haakonsson, Jesper G. S. Madsen, Mette Larsen, Isabel Forss, Martin R. Madsen, Elvira L. Van Hauwaert, Christian Wiwie, Naja Z. Jespersen, Michaela Tencerova, Ronni Nielsen, Bjørk D. Larsen, Richard Röttger, Jan Baumbach, Camilla Scheele, Moustapha Kassem & Susanne Mandrup (2019). Nature Genetics volume 51, pages 716–727. https://doi.org/10.1038/s41588-019-0359-1
In this work we have performed comprehensive characterization of the transcriptional and epigenomic changes associated with osteoblast and adipocyte differentiation of human MSC. We demonstrate that adipogenesis is driven by considerable remodeling of the chromatin landscape and de novo activation of enhancers, while osteogenesis involves activation of pre-established enhancers. Using machine learning algorithms for in silico modeling of transcriptional regulation we identify a large and diverse transcriptional network of pro-osteogenic and anti-adipogenic transcription factors. Intriguingly, binding motifs of these factors overlap with single nucleotide polymorphisms related to bone and fat formation in humans, and knockdown of single members of this network is sufficient to modulate differentiation in both directions, indicating that lineage-determination is a delicate balance between activities of many different transcription factors.
Integrated analysis of motif activity and gene expression changes of transcription factors
Jesper Grud Skat Madsen*, Alexander Rauch*, Elvira Laila Van Hauwaert, Søren Fisker Schmidt, Marc Winnefeld, Susanne Mandrup (2018). Genome Research 28, 243-255. https://doi.org/10.1101/gr.227231.117
In this study, we have developed a new and powerful computational tool, IMAGE, for precise prediction of transcription factors and target enhancers that are causally involved in driving specific transcriptional responses in time series or multi-group comparison studies, such as comparisons of cell types. IMAGE analyzes gene expression and enhancer activity data using machine learning to solve a model of transcriptional regulation, where the ‘activity’ of transcription factor motifs within enhancers drive gene expression in an additive and distance-dependent manner. Based on these motif activities, and gene expression patterns, IMAGE predicts causal transcriptional regulators. This approach offers several advantages over existing methods, including a more direct measure of motif importance leading to better predictions, as well as a more complete database of transcription factor motifs and a better scoring method, thereby reducing the bias in transcription factor prediction.
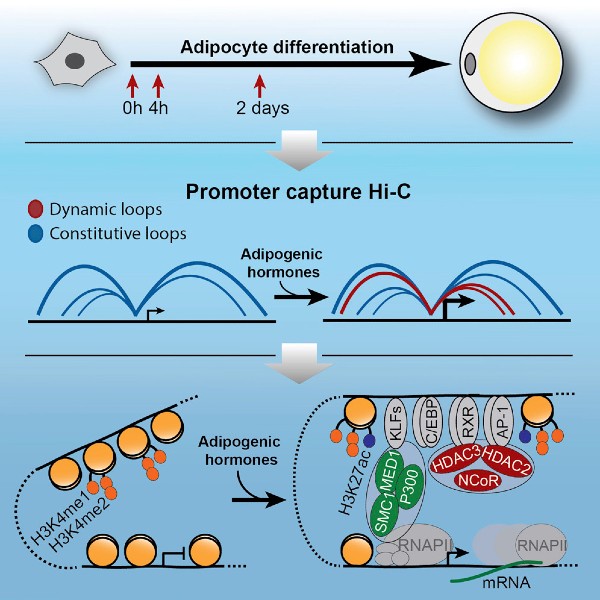
Dynamic Rewiring of Promoter-Anchored Chromatin Loops during Adipocyte Differentiation
Rasmus Siersbæk, Jesper Grud Skat Madsen, Biola Maria Javierre, Ronni Nielsen, Emilie Kristine Bagge, Jonathan Cairns, Steven William Wingett, Sofie Traynor, Mikhail Spivakov, Peter Fraser, Susanne Mandrup (2017). Molecular Cell 66, 420-435. https://dx.doi.org/10.1016/j.molcel.2017.04.010
In this study, we have used promoter capture Hi-C to for the first time demonstrate a rapid and dynamic rewiring of promoter-enhancer loops in response to differentiation stimuli. We show that rather than being pre-established promoter-anchored chromatin loops are dynamically rewired during early differentiation of 3T3-L1 preadipocytes. The establishment of new promoter-enhancer loops is tightly coupled to activation of enhancers, as evidenced by recruitment of co-activators and acquisition of active histone marks, as well as to activation of gene expression from the promoters. Intriguingly, formation of loops connecting activated enhancers and promoters is also associated with extensive recruitment of corepressors such as NCoR and HDACs, indicating that this class of coregulators may play a previously unrecognized role during enhancer activation.
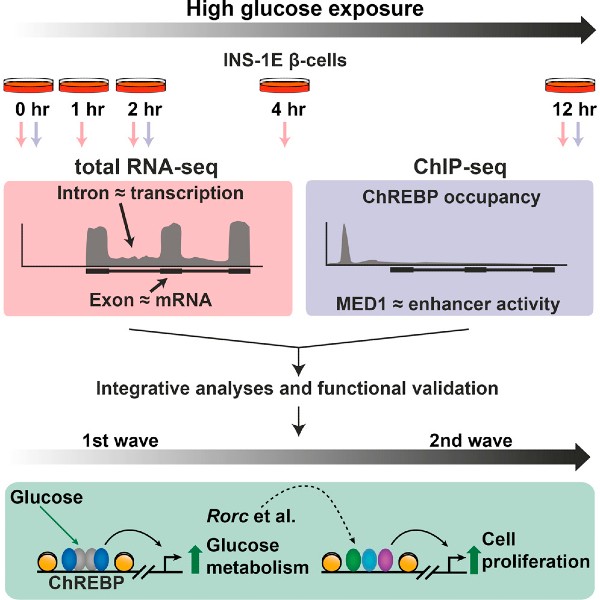
Integrative genomics outlines a biphasic glucose response and a ChREBP-RORgamma axis regulating proliferation in β-cells
S.F. Schmidt*, J.G.S. Madsen*, K. Østerli, L.L.C. Poulsen, Sofia Salö, M. Børgesen, A. Loft, R. Nielsen, J.J.Holst, Louise T. Dalgaard, S. Mandrup (2016). Cell Reports 16, 1-14. http://dx.doi.org/10.1016/j.celrep.2016.07.063
In this paper, we used a detailed functional genomic and transcriptomic mapping strategy to characterize the glucose response in pancreatic β-cells. We demonstrate that the glucose response is biphasic. During the first wave, it is predominantly the gene expression of metabolic enzymes that is regulated. However, during the second wave, there is induction of genes involved in cell cycle and repression of key phenotype-defining genes, such as insulin 1. Carbohydrate response element binding protein (ChREBP), a glucose-activated transcription factor, orchestrates the entire transcriptional program by directly inducing first wave genes, and the transcription factors responsible for the regulation of second wave genes. One of these transcription factors is the RAR-related orphan receptor gamma (RORγ). We show that knockdown of RORγ blunts both the glucose-mediated induction of cell cycle genes, as well as the glucose-mediated induction of proliferation. Collectively, these findings constitute a detailed dissection of the transcriptional response to glucose in pancreatic β-cells, and identify novel regulators of glucose-induced proliferation.
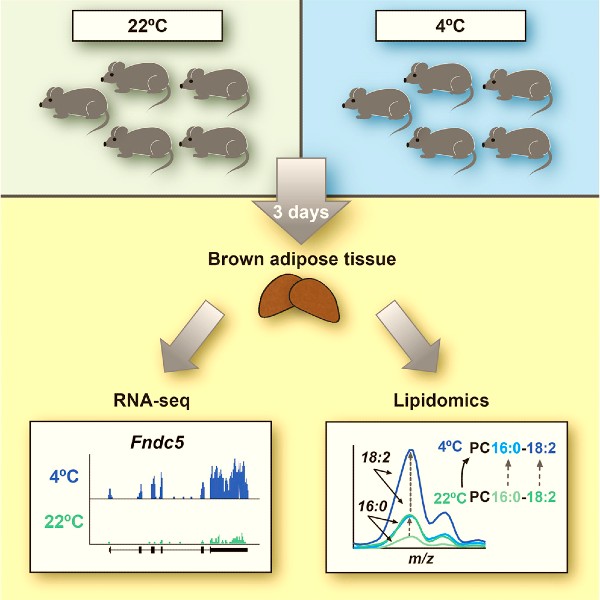
RNA-Seq and Mass-Spectrometry-Based Lipidomics Reveal Extensive Changes of Glycerolipid Pathways in Brown Adipose Tissue in Response to Cold
Ann-Britt Marcher, Anne Loft, Ronni Nielsen, Terhi Vihervaara, Jesper Grud Skat Madsen, Marko Sysi-Aho, Kim Ekroos, Susanne Mandrup (2015).Cell Reports 13, 2000–2013. http://dx.doi.org/10.1016/j.celrep.2015.10.069
In this study, we combined RNA sequencing and mass spectrometry-based lipidomics and provide a comprehensive resource that describes the molecular signature of cold adaptation of the brown adipose tissue (BAT) to cold, at the level of the transcriptome and lipidome. Upon short-term (3-days) cold exposure we find a robust induction of several brown adipocyte genes related to thermogenesis, as well as the gene encoding the hormone Irisin. However, the most significantly induced gene programs relate to glycerophospholipid synthesis and fatty acid elongation, which is accompanied by significant changes in the acyl chain composition of triacylglycerols (TAGs) and subspecies-selective changes in the acyl chains in glycerophospholipids. In particular, long-chain and odd-numbered acyl chains in TAGs are increased in response to cold. These findings indicated that the cold adaptation of BAT is associated with significant and highly species-selective remodeling of both TAGs and glycerophospholipid species.
Acute TNF-induced repression of cell identity genes is mediated by NFκB-directed redistribution of cofactors from super-enhancers
Søren Fisker Schmidt, Bjørk Ditlev Larsen, Anne Loft, Ronni Nielsen, Jesper Grud Skat Madsen, Susanne Mandrup (2015). Genome Research. http://dx.doi.org/10.1101/gr.188300.114
In this paper we demonstrate that the major transactivating subunit of NFκB, RELA, is also required for acute TNF-induced suppression of adipocyte genes. Notably, this repression does not involve RELA binding to the associated enhancers but rather loss of cofactors and enhancer RNA selectively from high occupancy sites within super-enhancers. Our observations, which also apply to other cell types, propose a novel paradigm for NFκB-mediated repression, whereby NFκB selectively redistributes cofactors from high occupancy enhancers, thereby specifically repressing super-enhancer-associated cell identity genes.
iRNA-seq: computational method for genome-wide assessment of acute transcriptional regulation from total RNA-seq data
Jesper Grud Skat Madsen, Søren Fisker Schmidt, Bjørk Ditlev Larsen, Anne Loft, Ronni Nielsen, Susanne Mandrup (2015). Nucl. Acids Res. http://dx.doi.org/10.1093/nar/gku1365
In this paper, we developed a computational method (iRNA-seq) to assess acute transcriptional change using introns and total RNA-seq data and provided and easy-to-use command line tool. As proof-of-principle, we compared the results of iRNA-seq to both RNA Polymerase II ChIP-seq and mRNA-based RNA-seq analysis of transcriptional changed in TNF treated SGBS adipocytes. We show that iRNA-seq is substantially more sensitive than mRNA-based analysis and comparable to that of RNA Polymerase II ChIP-seq for acute transcriptional changes. Furthermore, using publicly available datasets, we show that the results of iRNA-seq strongly correlate with those of GRO-seq. In summary, this paper demonstrates that total RNA-seq and iRNA-seq analysis is approximately as sensitive as both RNA Polymerase II ChIP-seq and GRO-seq, while being much faster, easier and requiring a lot less material.
Browning of human adipocytes requires KLF11 and reprogramming of PPARγ superenhancers
Loft A, Forss I, Siersbæk MS, Schmidt SF, Larsen AS, Madsen JG, Pisani DF, Nielsen R, Aagaard MM, Mathison A, Neville MJ, Urritia R, Karpe F, Amri EZ, Mandrup S (2015). Genes & Development, 29, 7-22. http://dx.doi.org/10.1101/gad.250829.114
In this study, we characterized the transcriptional processes underlying browning of human adipocytes using a combination of RNA-, ChIP-, and DHS-seq analyses. As a model system we used human multipotent adipose-derived stem (hMADS) adipocytes exposed for 3 days to the synthetic PPARγ agonist, rosiglitazone, which stimulate the formation of functional brown in white (i.e. brite) adipocytes. We demonstrated that browning activates a comprehensive gene program associated with increased mitochondrial function and energy consumption. Interestingly, the browning process involves reprogramming of PPARγ binding to form brite-selective PPARγ super-enhancers that appear to play a key role in activation of the brite-selective gene program, including putative regulators of the browning process. Based on its association with a PPARγ super-enhancer we identify KLF11 as a novel browning factor directly induced by rosiglitazone and required for the activation of the brite-selective gene program in human adipocytes. Cumulatively, this study provides novel mechanistic insight into the transdifferentiation from white to brite fat cells.
There was a lot of media coverage of this publication. Click here to see more (in Danish).
Transcription factor cooperativity in early adipogenic hotspots and super-enhancers
R. Siersbæk, A. Rabiee, R. Nielsen, S. Sidoli, A. Loft, L.L.C. Poulsen, A. Rogowska-Wrzesinska, O.N. Jensen, S. Mandrup (2014) Cell Reports, 7, 1443–1455. http://dx.doi.org/10.1016/j.celrep.2014.04.042
In this paper, we use a combination of advanced genomics and proteomics approaches to identify transcriptional regulators that are part of the early adipogenic transcription factor network. Based on ChIP-seq profiling of many of the identified transcription factors, we demonstrate that transcription factors tend to co-localize in specific hotspots of the genome that have all the characteristics of enhancers. Interestingly, these hotspots are highly enriched in large super-enhancer regions characterized by ultra-high densities of transcription factors and co-activators, and these super-enhancers appear to be very important regulators of gene activation. Importantly, we demonstrate extensive transcription factor cooperativity at the level of hotspots as well as super-enhancers, which is important for enhancer activity and gene activation. Collectively, this demonstrates that transcriptional regulators cooperate extensively at specific genomic hubs to reprogram gene expression.
Molecular architecture of transcription factor hotspots in early adipogenesis
R. Siersbæk, S. Baek, A. Rabiee, R. Nielsen, S. Traynor, N. Clark, A. Sandelin, O.N. Jensen, M.-H. Sung, G.L. Hager, S. Mandrup (2014) Cell Reports, 7, 1434–1442. http://dx.doi.org/10.1016/j.celrep.2014.04.043
In this report, we provide molecular insight into the organization of transcription factors at adipogenic transcription factor hotspot regions using digital genomic footprinting. We demonstrate for the first time in a chromatin context that C/EBP-ATF heterodimers form on a composite motif. Furthermore, we show that transcription factors are extensively recruited to hotspots through alternative mechanisms that do not involve their known DNA binding motif. The recruitment of factors through alternative mechanisms is usually associated with direct binding of other factors to DNA (e.g. C/EBPs, AP1, and KLFs). Interestingly, recruitment of transcription factors through alternative mechanisms is functionally important for hotspot formation, enhancer activity, and gene regulation. Taken together, these data provide a new framework for understanding transcription factor cooperativity at hotspot regions.
Peroxisome proliferator activated receptor γ and C/EBPα synergistically activate key metabolic adipocyte genes by assisted loading.
M.S. Madsen, R. Siersbæk, M. Børgesen, R. Nielsen, S. Mandrup (2014) Mol. Cell. Biol. 34, 939-954. http://dx.doi.org/10.1128/MCB.01344-13
In this publication, we provide molecular insight into the cross-talk between the key transcriptional regulators of adipogenesis, PPARγ and C/EBPα, at shared binding sites. We established a fibroblastic cell model system that allowed us to independently control the expression of PPARγ and C/EBPα and investigate the acute effects of these factors on gene regulation. RNA-seq analyses revealed that co-expression of both factors resulted in synergistic activation of several key adipocyte genes that are also activated during 3T3-L1 adipogenesis. These synergistic genes were associated with C/EBPα-mediated reprogramming of PPARγ binding and vice versa. Subsequent mechanistic analyses revealed that an assisted loading mechanism involving chromatin remodeling by the leading factor could explain many of these reprogramming events. Taken together, this provides novel mechanistic insight into the cross-talk between PPARγ and C/EBPα on chromatin, which is important for regulation of the adipocyte gene program.
Delayed hepatic adaptation to weaning in ACBP-/- mice is caused by disruption of the epidermal barrier
D. Neess, S. Bek, A.-B. Marcher, M. Bloksgaard, N. Færgeman, S. Mandrup(2013) Cell Reports 5, 1403-1412. http://dx.doi.org/10.1016/j.celrep.2013.11.010
In this publication we report a link between epidermal barrier function and hepatic gene regulation and metabolism. Impaired epidermal barrier function through skin-specific ACBP disruption leads to suppression of the hepatic lipogenic gene program during weaning. This phenotype can be rescued by applying an artificial skin barrier. The data support a model in which increased lipolysis in white adipose tissue results in hepatic accumulation of lipids and suppression of hepatic SREBP activity.
Genome-wide profiling of PPARγ in primary epididymal, inguinal and brown adipocytes reveals depot-selective binding correlated with gene expression
M.S. Siersbæk*, A. Loft*, M. M. Aagaard*, R. Nielsen, S. F. Schmidt, N. Petrovic, J. Nedergaard, S. Mandrup(2012) Mol. Cell Biol. 32, 3452–3463. (Selected for spotlight). http://dx.doi.org/10.1128/MCB.00526-12
In this publication we have used chromatin immunoprecipitation combined with deep sequencing (ChIP-seq) to map the genome-wide binding profile of the master regulator of adipocyte differentiation, peroxisome proliferator-activated receptor γ (PPARγ), in different types of in vitro differentiated primary mouse adipocytes. This allows us for the first time map PPARγ binding in primary mouse adipocytes and to investigate how binding patterns differ between brown and white adipocytes and between different types of white adipocytes. Taken together, our study indicates that differential preprogramming of preadipocytes in the different adipose depots facilitates depot-selective PPARγ recruitment to target sites, and that PPARγ binding to these sites plays a role in depot-specific gene expression.
The acyl coenzyme A binding protein is required for epidermal barrier function in mice.
M. Bloksgaard, S. Bek, A.-B. Marcher, D. Neess, J. Brewer, H.K. Hannibal-Bach, T. Helledie, C. Fenger, M. Due, Z. Berzina, R. Neubert, J. Chemnitz, B. Finsen, A. Clemmensen, J. Wilbertz, H. Saxtorph, J. Knudsen, L. Bagatolli, S. Mandrup (2012) J. Lipid Res. 53, 2162-74. http://dx.doi.org/10.1194/jlr.M029553
In this report on we show that mice with targeted disruption ACBP display a clearly visible skin and fur phenotype characterized by greasy fur and development of alopecia and scaling with age as well as a 50% increased water loss over the epidermal barrier. Biochemically, we demonstrate a significant decrease in the levels of very long chain fatty acids in stratum corneum of ACBP-/- mice, which is likely to play an important causal role in the decrease in epidermal barrier function.
Genome-wide profiling of LXR, RXR and PPARα in mouse liver reveals extensive sharing of binding sites
M. Børgesen, T. Å. Pedersen, S. vanHeeringen, B. Gross, Hagenbeek, C. Bindesbøll, S. Caron, F. Lalloyer, K. Steffensen, H. Nebb, J.-Å. Gustafsson, H. Stunnenberg, B. Staels, S. Mandrup (2012) Mol Cell Biol. 32, 852-867. http://dx.doi.org/10.1128/MCB.06175-11
In this publication we have used chromatin immunoprecipitation combined with deep sequencing (ChIP-seq) to map and investigate the liver X receptor (LXR): retinoid X receptor (RXR) transcriptional network in mouse liver. The work represents the first genome-wide profile of LXR, RXR and PPAR binding sites in liver and the first to report the effect of agonist on LXR and RXR receptor occupancy at a genome-wide level. Furthermore, we report the surprising discovery that there is an extensive overlap between the LXR and PPARα binding, and we provide evidence indicating that these receptors bind in mutually exclusive manner to shared binding sites.
Extensive chromatin remodeling and establishment of transcription factor ‘hotspots’ during early adipogenesis
R. Siersbæk, R. Nielsen, S. John, M.-H. Sung, S. Baek, A. Loft, G. L. Hager¤, S. Mandrup¤ (2011) EMBO J 30, 1459-72 (highlighted in ‘Have You Seen’ in the same issue by D. Steger and M.A. Lazar) (¤ co-correspondence). http://dx.doi.org/10.1038/emboj.2011.65
In this publication we employed DNase hypersentivity (DHS) site analysis in combination with deep sequencing (DHS-seq) to obtain a temporal genome-wide map of the chromatin structure during adipocyte differentiation. This revealed a dramatic induction of new DHS sites early in differentiation. Bioinformatic analysis of the DNA sequences underlying the DHS sites was used to predict candidate transcription factors involved in the remodeling. Interestingly, the genome-wide profiles of these candidate factors revealed >1000 transcription factor ‘hotspots’ that are co-occupied by multiple transcription factors early in adipogenesis. This work represents the first genome-wide map of chromatin transitions through a differentiation process and reveals a novel crosstalk between early and late events during adipocyte differentiation. The paper was highlighted in “Have You Seen” in the same issue.
ChREBP mediates glucose-repression of PPARα expression in pancreatic β-cells
M. Børgesen*, L.L.C. Poulsen*, S. F. Schmidt, F. Frigerio, P. Maechler, S. Mandrup (2011) J. Biol. Chem. 286, 13214-13225 (*equal contribution). http://dx.doi.org/10.1074/jbc.M110.215467
Using gain-of-function as well as loss-of-function approaches we show that ChREBP is a critical and direct novel mediator of glucose repression of PPARα in pancreatic β-cells. Our results indicate that glucose activation of ChREBP affects lipid partitioning in β-cells not only by activating the lipogenic gene program but also by repressing fatty acid oxidation through repression of PPARα and by stimulating expression of lipogenic genes, thereby suppressing fatty acid oxidation and inducing lipogenic capacity, respectively. Furthermore, the data suggest that ChREBP, which has primarily been implicated in glucose-induced activation of gene expression, may play an important and hitherto unrecognized role in glucose repression of gene expression in pancreatic β-cells.
Disruption of the acyl-CoA binding protein results in delayed hepatic adaptation to the metabolic changes at weaning
D. Neess*, M. Bloksgaard*, S. Bek, A.-B. Marcher, I.C. Elle, T. Helledie, M. Due, V. Pagmantidis, B. Finsen, J. Wilbertz, M. Kruhøffer, N. Færgeman, S. Mandrup (2011) J. Biol. Chem. 286, 3460-3472. (*equal contribution). http://dx.doi.org/10.1074/jbc.M110.161109
We have as the first generated mice with general targeted disruption of the acyl-CoA binding protein (ACBP), a small highly conserved lipid binding protein. These mice a viable and fertile but go through a crisis at weaning with increased mortality. In this report we show ACBP-/- mice display a delayed adaptation to weaning with significantly delayed induction of the hepatic lipogenic gene program. Our data indicate that this is due to increased lipid accumulation in the liver prior to weaning.
Genome-wide profiling of PPARγ:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis
R. Nielsen*, T. Å. Pedersen*, D. Hagenbeek*, P. Moulos, R. Siersbæk, E. Megens, M. Børgesen, K.-J. Francoijs, S. Mandrup¤ and H. G. Stunnenberg¤ (2008) Genes & Dev. 22, 2953 -2967. (* equal contribution, ¤ co-correspondence).http://dx.doi.org/10.1101/gad.501108
This paper reports the first genome profiles of transcription factors in adipocytes using chromatin immunoprecipitation in combination with deep sequencing (ChIP-seq). We demonstrate that PPARγ, which is considered the master regulator of adipocyte differentiation, is associated with the majority of genes upregulated (as determined by RNApolII ChIP-seq) during adipocyte differentiation. This indicates a much more general role of PPARγ in induction of adipocyte gene expression than previously anticipated. The heterodimerization partner of PPARγ, RXR, is associated with multiple target sites already prior to expression of PPARγ indicating a change of partner during differentiation.